

Basic Information
What should be noticed is, as the program usually takes more than 5 minutes, thus a valid email is required here, in case you forgot the link. If the program runs very well, we will send you an email later.
Identifier
Identifier is just the name of your program. You can give a name as to your own preference, like 'Jim's awesome run', 'My first run', '1122233', so on. When you click the 'Monitor Progress' button, you can recognize your program by your specific identifier.
Input file or paste variant
This is the place where you should submit your variant calls, either by a file or through paste.
Five different types of formats could be accepted here, separately, vcf, tsv, annovar, gff3 and masterVar.
The details of these formats could be seen through the Zoom In icon in the Parameter Setting section.
Phenotype/Disease Specific Prioritization (Call Phenolyzer)
Overview
Phenolyzer stands for Phenotype Based Gene Analyzer, a new software developed by us, which can prioritize genes based on specific diseases or phenotypes.
Call Phenolyzer is a pipeline connecting wANNOVAR with Phenolyzer, discovering genes directly from wANNOVAR output.
It is extremely helpful if the user provides some short disease/phenotype terms together with the variant file,
then robust ontology searching and machine learning method is used to discover the candidate genes.
To see more Phenolyzer examples, please click this link
To see the FAQ of Phenolyzer, please click this link
Enter Phenotypes
Enter Phenotypes or Diseases here. Please enter some terms here if you already have an idea about which disease/phenotype you
are interested in or already observed. Several kinds of terms are accepted here, including:
1) disease names: like alzheimer, parkinson, chron, and so on;
2) phenotype names: like fatigure, Blood pressure. Any HPO (Human Phenotype Ontology) term is also accepted.
3) OMIM IDs: like 114480 for'breast cancer'.
Please use Enter or ';' as separators, some other non-word characters are also acepted like '|'.
Please try to use short terms instead a very long term. As the first step of Phenolyzer is to do word match between your
word and the names in our database. Thus super long names might not have any results.
For example, 'X-linked intellectual deficiency' is better to be separated into
'X-linked' and 'intellectual deficiency'.
Understand Results
If any Phenotype/Disease terms are entered, there will be an addtional section in the result, named 'Phenotype/disease Prioritization Result'.
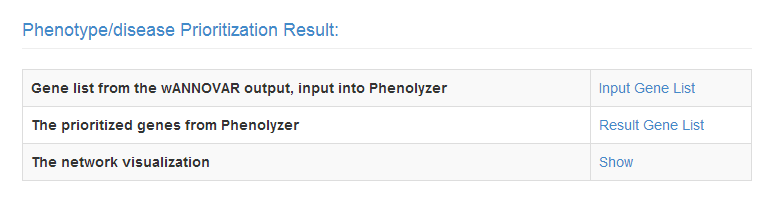
The first row 'Input Gene List' is the gene list extracted from the ANNOVAR output variant calling list.
Tip: After your first run of wANNOVAR, you can run Phenotype/Disease specific prioritization with some other terms without the need to run wANNOVAR, which is
more time and computation efficient as Phenolyzer could finish in one minute and wANNOVAR usuually takes more than 5 minutes. What you need to do is just
take the Input Gene List here, go to Phenolyzer website and submit it and your new phenotypes there!
'Result Gene List' is the output from Phenolyzer, with the information of ranks, gene names, NCBI IDs, and scores.
'Show' will lead you to the Phenolyzer's network visualization. You could do interactive research there, like finding all
the genes and diseases interacting with one gene by double click the gene. You can also see the details about how one gene is discovered
through the prioritization algorithm.
Parameter Setting
This part is mainly for advanced settings of wANNOVAR. You can change the genome build, input format, gene definition, and disease filter here.
The details of them could be found by clicking the 'Zoom in' button following each input element.
NOTICE: If you use the custom filtering option, please at least choose a filter step in
the section below, or use a control file.
Reference Genome
- hg18:The Homo Sapiens hg18 genome assemblyhttps://genome.ucsc.edu/cgi-bin/hgGateway?db=hg18
- hg19:The Homo Sapiens hg19 genome assemblyhttps://genome.ucsc.edu/cgi-bin/hgGateway?db=hg19
Input Format
- VCF genotype calling format: This is the most widely used format for exchanging variant calls. Details can be found on the 1000 Genomes web site:
http://www.1000genomes.org/wiki/Analysis/Variant%20Call%20Format/vcf-variant-call-format-version-41. - ANNOVAR input format: This is the standard file format used by the ANNOVAR software. Details can be found on at:
http://www.openbioinformatics.org/annovar/annovar_input.html. - Complete Genomics format: the var-ASM.TSV file generated by Complete Genomics standard pipeline. Future versions of wANNOVAR will also support the masterVarBeta-ASM.TSV format. More details can be found at: http://media.completegenomics.com/documents/DataFileFormats+Standard+Pipeline+2.0.pdf.
- GFF3-SOLiD input format: The GFF file generated from SOLiD sequencer is a text based repository and contains data and analysis results; colorspace calls, quality values (QV) and variant annotations. See details at http://www3.appliedbiosystems.com/cms/groups/mcb_marketing/documents/generaldocuments/cms_058718.pdf.
- Complete Genomics masterVar format: This is the file format (masterVarBeta-ASM.TSV format) used by Complete Genomics for all kinds of variation data and can be used to analyze and visualize the variant calls made by Complete Genomics.
Gene Definition
- RefSeq Gene: Gene annotation released by The Reference Sequence (RefSeq) database, which is an open access, annotated and curated collection of publicly available nucleotide sequences (DNA, RNA) and their protein products. This database is built by National Center for Biotechnology Information (NCBI), and, unlike GenBank, provides only single record for each natural biological molecule(i.e. DNA, RNA or protein) for major organisms ranging from viruses to bacteria to eukaryotes.
- UCSC Known Gene: Gene annotation released by The University of California Santa Cruz (UCSC) Known Genes dataset, which is constructed by a fully automated process, based on protein data from Swiss-Prot/TrEMBL (UniProt) and the associated mRNA data from Genbank.
- Ensembl Genes: In the Ensembl project, sequence data is fed into a software "pipeline" to creates a set of predicted gene locations.
- GENCODE Genes: The GENCODE genes are compiled by a combination of initial manual annotation and experimental validation by the GENCODE consortium, as well as a refinement of the annotation based on these experimental results.
Disease Model
- rare recessive Mendelian disease: More than two deleterious alleles (compound heterozygote or homozygote or hemizygote in chrX in males) are found in the same gene. wANNOVAR will identify splicing variants and exonic variants that change protein coding, remove variants observed in 1000 Genomes Project, NHLBI-ESP 5400 exomes and dbSNP, and identify a list of candidate genes.
- rare dominant Mendelian disease: More than one deleterious allele is found in the gene. wANNOVAR will identify splicing variants and exonic variants that change protein coding, remove variants observed in 1000 Genomes Project, NHLBI-ESP 5400 exomes and dbSNP, and identify a list of candidate genes.
- X-linked recessive (Male): The same as rare dominant, variants should be in X chr of a male.
- X-linked recessive (Female): The same as rare recessive, variants should be in X chr of a female.
- X-linked dominant: The same as rare dominant, variants should be in X chr.
- custom filtering: Users specify customized variants reduction criteria in "advanced options".
Custom Filtering
- Depth threshold (VCF only): Specify read depth threshold.
- Quality threshold (VCF only): Specify quality score threshold.
- MAF in variants filtering: Specify Minor allele frequency threshold.
- dbSNP version: Specify dbSNP version for filtering.
- disease type: Activated When "custom filtering" option is selected. Identify candidate genes with more than two deleterious alleles (compound heterozygote or homozygote or hemizygote in chrX in males) are found in the same gene for "recessive". Identify candidate genes with more than one deleterious allele is found in the gene for "dominant".
- Variants filtering: Activated When "custom filtering" option is selected. Specify the optimized filtering step according to users' samples.
- Variant file on controls: A variants list file is required when "not in controls" step is selected in "Variants filtering pipeline". wANNOVAR will remove the candidate variants which are also occured in controls.
- Paste variants on controls: Paste the variants list on controls directly.
Table below lists the some columns of variants annotation from wANNOVAR. For other details, please see ANNOVAR website: region based annotation and filter based annotation.
| Column Name | Explanation |
|---|---|
| Func | Variant function (exonic, intronic, intergenic, UTR, etc) |
| Gene | Gene Name. By default, RefSeq gene definition is used, but users can choose from other gene definition systems. |
| ExonicFunc | Exonic variant function (non-synonymous, synonymous, etc) |
| AAChange | Amino acid changes |
| Conserved | Region-level phastCons LOD scores |
Version Information
wANNOVAR uses ANNOVAR for annotation, and its corresponding database version is given in this section. 1000G: 2015aug; ExAC: exac03; esp: esp6500siv2; dbSNP: avsnp147; cosmic: cosmic81; clinvar: 20170501; gwas: avgwas_20170516; dbNSFP (scores for missense variants): dbnsfp33a; gnomad: gnomad_exome, gnomad_genome. For more details on these databases, please check the ANNOVAR website.